
未来生命科学開拓部門に所属する本研究グループは、iPS細胞技術をツールとして活用し、分子細胞レベルでの新たな生命科学の分野を開拓する研究を実施します。特に、iPS細胞誘導過程を含む細胞運命変換過程や個体の老化過程を対象実験系として用い、生命の設計図であるゲノムDNAからどのように情報が取り出され、取り出された情報がどのように細胞の機能や性質へと結びつくのかを明らかにすることによって、生命現象の根本原理を見出すことを目指します。本研究グループでは、分子生物学、生化学、細胞生物学、遺伝学、網羅的解析技術、バイオインフォマティクス、等を駆使し、ゲノムDNAから写し取られたRNAの発現制御や機能制御といった多階層に跨がる制御機構を統合的に解析することによって、上記の目標達成にチャレンジします。多階層に跨がる制御機構として、染色体高次構造制御、エピジェネティック制御、転写制御、転写後修飾制御、RNA2次構造制御、翻訳制御を含み、それぞれの制御層間の関連性の解明も研究課題です。
Our group aims to discover the basic principles underlying biological phenomena by elucidating the molecular mechanisms that process cellular information (genomic DNA) into cellular properties and functions. We are studying cell fate conversion processes, including those in iPSC induction and organism aging, as research targets. We study these processes by an integrative analysis of multi-hierarchical DNA and RNA regulatory systems. Our methods include molecular biology, biochemistry, cell biology, genetics, genome-wide analyses and bioinformatics techniques.
Research content
研究内容
Higher-order chromatin structures
染色体高次構造制御
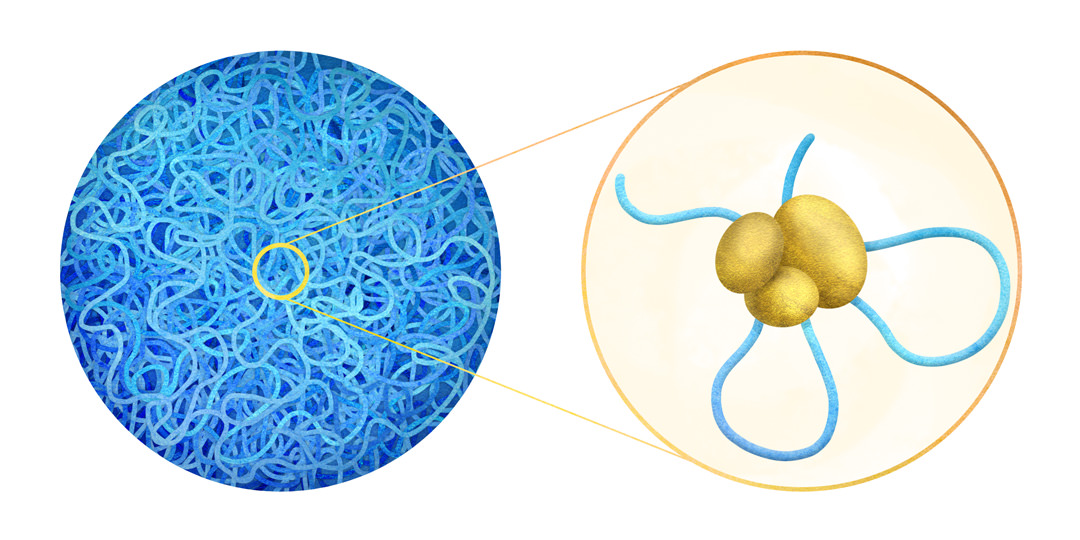
ゲノム情報を完全に理解するためには、ゲノムDNAの1次配列だけではなく、3次元空間上での配置を知る必要があります。私たちの研究グループは、数十のゲノム領域の染色体相互作用を高精度で同定できるms4C-seqを開発しました。ms4C-seqで得た相互作用情報と染色体の核内配置に関するデータとの統合的解析により、多能性幹細胞において発生関連遺伝子座は核の内部で集合しやすい傾向にあること、体細胞初期化過程において発生関連遺伝子座が再集合すること、それら遺伝子座の再集合にはエピジェネティック制御因子が必要であることを見出しました (Ikeda et al., Nature Communications 2017) 。現在、ms4C-seqを用いて、遺伝子砂漠上において、多能性幹細胞で重要な染色体高次構造の同定を試みています。
To understand genomic information comprehensively, we should know not only the primary genomic DNA sequences, but also the arrangements of these sequences in the three-dimensional space of the nucleus. Our group has developed ms4C-seq, a technique that can identify chromatin interactions with high accuracy at several tens of genomic loci simultaneously (Ikeda et al., Nature Communications 2017). By integrative analyses of the chromatin interaction data obtained from ms4C-seq and chromatin nuclear arrangement data, we have demonstrated that developmental gene loci tend to colocalize in pluripotent stem cells (PSCs), and that this colocalization requires epigenetic modifiers which are essential regulatory factors for the maintenance of transcriptionally poised developmental genes (Ikeda et al., Nature Communications 2017). Now, we are trying to identify crucial chromatin interactions for PSCs in gene desert regions by using ms4C-seq.
Epigenetic regulation
エピジェネティック制御

本研究グループは、ヒストン修飾やDNAメチル化といったエピジェネティック制御機構の解析を行なってきました (Yamashiro et al., Science 2018, Ohnishi et al., Cell 2014)。例えば、山田泰広教授(現東京大学医科学研究所)との共同研究で、アレル特異的なDNAメチル化解析を行い、インプリンティング状態を網羅的かつ精度良く調べることによって、ゲノムインプリンティングを維持した高い個体発生能をもつES細胞を効率よく樹立することに成功しました (Yagi et al., Nature 2017)。現在、iPS細胞樹立とゲノムインプリンティングに関する研究やiPS細胞を用いたDNAメチル化機構解明についての研究を行なっています。
Our group has performed analyzed several aspects of epigenetic regulation including histone modification and DNA methylation (Yamashiro et al., Science 2018, Ohnishi et al., Cell 2014). For example, in collaboration with Prof. Yamada (Tokyo University), by performing allele-specific comprehensive DNA methylation analysis, we have succeeded in deriving ground-state female ESCs while maintaining gamete-derived DNA methylation (Yagi et al., Nature 2017). Now, we are studying genome imprinting regulation during somatic cell reprogramming and the DNA methylation mechanism in iPSCs.
Transcriptional regulation
転写制御
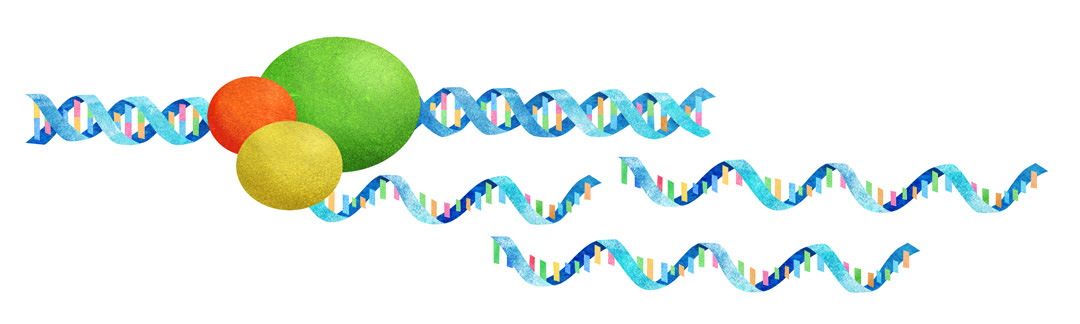
本研究グループは、転写因子EsrrbとZic3という2つの転写因子を山中ファクターと同時に体細胞に導入するとマウスの体細胞初期化効率を10倍以上増加させることを明らかにしました。それら2つの転写因子の詳細な機能解析により、多能性転写ネットワークが、代謝ネットワークの恒常性をバランス良く協調的に変化させることによって、細胞の運命を変換させることを示しました (Sone et al., Cell Metabolism 2017)。
We have shown that when transduced with the Yamanaka factors into murine fibroblasts, two transcription factors (TFs), Zic3 and Esrrb, synergistically enhance the reprogramming efficiency by more than one order of magnitude compared to the Yamanaka factors alone (Sone et al., Cell Metabolism 2017). Further, we discovered that these TFs cooperatively activate glycolytic metabolism during the reprogramming. At the same time, they antagonistically regulate OXPHOS, with Zic3 repressing OXPHOS and Esrrb activating it. Therefore, when introduced with Zic3, Esrrb restores OXPHOS activity, a phenomenon we found essential for efficient reprogramming. Overall, this study suggests that the combinatorial function of TFs achieves an appropriate balance of metabolic pathways to induce naive PSCs.
Post-transcriptional regulation
転写後修飾制御

本研究グループは、トランスクリプトーム解析によって得られたデータを利用し、網羅的選択的スプライシング解析を行ないました。結果、スプライシングパターンは体細胞初期化過程において体細胞様からES細胞用様にダイナミックに変化すること、それらスプライシングパターンの変化は初期化のある特定の時期で一斉に起こるのではなく、遺伝子毎に異なったタイミングで起こること等を示しました。また、siRNAスクリーニング等を用いることにより、U2af1とSrsf3というRNA結合タンパク質が多能性幹細胞のスプライシングを制御し、体細胞初期化過程でも重要な役割を果たしていることを見出しました (Ohta et al., Cell Reports 2013)。また、RNAの2次構造を網羅的に同定する手法により、RNA2次構造と種々の転写後修飾の関係性やRNA2次構造が翻訳制御に与える影響を調べています。
We have performed transcriptome analysis and identified more than 500 genes whose splicing patterns are changed during somatic cell reprogramming (Ohta et al., Cell Reports 2013). We also showed that somatic splicing profiles revert to pluripotent ones during reprogramming and that changes in the splicing pattern occur stepwise at particular stages in reprogramming. Furthermore, our siRNA screening and biochemical experiments demonstrated that two RNA-binding proteins, U2af1 and Srsf3, play a role in somatic cell reprogramming. Now, by using methods to identify RNA secondary structures comprehensively, we are studying the relationship between RNA secondary structures and post-transcriptional regulation and the effects of RNA secondary structures on protein translational regulation.
Translational regulation
翻訳制御

山中伸弥教授(京都大学iPS細胞研究所)との共同研究で、本研究グループで行なったリボソームプロファイリング手法により、翻訳開始因子のひとつであるNAT1ノックアウト細胞で、翻訳制御が変化する遺伝子群を同定し、マウス多能性幹細胞におけるNAT1の翻訳制御機構の一端を解明しました (Sugiyama et al., Proc. Natl. Acad. Sci. U. S. A. 2017) 。
In collaboration with Prof. Yamanaka (CiRA), we performed a ribosomal profiling that helped identify genes whose translation differs in normal and NAT1 (a translation-initiation factor) knockout ESCs and elucidated the mechanism for how NAT1 mediates protein translation in mouse PSCs (Sugiyama et al., Pro. Natl. Acad. Sci. U. S. A. 2017).
Main equipment
主要設備 (共通機器)

-
HiSeq2500, cBot
変異解析、RNA2次構造解析、バイサルファイトシーケンスを用いたDNAメチル化解析等に使用してます。
This equipment is for the analyses of mutations, RNA secondary structures, DNA methylation, etc.

-
NextSeq 500
RNA-seq、SC3-seq 、ChIP-seq、ATAC-seq、iCLIP、リボソームプロファイリング、等に使用しています。
This equipment is for RNA-seq, SC3-seq, ChIP-seq, ATAC-seq, iCLIP, ribosome profiling, etc.

-
MiSeq
Amplicon-seq, TargetRNA-seq, smallRNA-seq, ライブラリのQC等に使用しています。
This equipment is for Amplicon-seq, TargetRNA-seq, smallRNA-seq, library QC, etc.